Intro
0:00Organic Synthesis Strategies
0:15Goal
0:16Strategy
0:29
Example of a RetroSynthesis
1:30Finding Starting Materials for Target Molecule
1:31Synthesis Using Starting Materials
4:56
Synthesis of Alcohols by Functional Group Interconversion (FGI)
6:00Synthesis of Alcohols by Functional Group Interconversion Overview
6:01
Alcohols by Reduction
7:43Ketone to Alcohols
7:45Aldehyde to Alcohols
8:26Carboxylic Acid Derivative to Alcohols
8:36
Alcohols by Hydration of Alkenes
9:28Hydration of Alkenes Using H₃O⁺
9:29Oxymercuration-Demercuration
10:35Hydroboration Oxidation
11:02
Alcohols by Substitution
11:42Primary Alkyl Halide to Alcohols Using NaOH
11:43Secondary Alkyl Halide to Alcohols Using Sodium Acetate
13:07Tertiary Alkyl Halide to Alcohols Using H₂O
15:08
Synthesis of Alcohols by Forming a New C-C Bond
15:47Recall: Alcohol & RMgBr
15:48Retrosynthesis
17:28
Other Alcohol Disconnections
19:46
19:47Synthesis Using PhMGgBr: Example 2
23:05
Synthesis of Alkyl Halides
26:06Synthesis of Alkyl Halides Overview
26:07
Synthesis of Alkyl Halides by Free Radical Halogenation
27:04Synthesis of Alkyl Halides by Free Radical Halogenation
27:05
Synthesis of Alkyl Halides by Substitution
29:06Alcohol to Alkyl Halides Using HBr or HCl
29:07Alcohol to Alkyl Halides Using SOCl₂
30:57Alcohol to Alkyl Halides Using PBr₃ and Using P, I₂
31:03
Synthesis of Alkyl Halides by Addition
32:02Alkene to Alkyl Halides Using HBr
32:03Alkene to Alkyl Halides Using HBr & ROOR (Peroxides)
32:35
Example: Synthesis of Alkyl Halide
34:18Example: Synthesis of Alkyl Halide
34:19
Synthesis of Ethers
39:25Synthesis of Ethers
39:26
Example: Synthesis of an Ether
41:12Synthesize TBME (t-butyl methyl ether) from Alcohol Starting Materials
41:13
Synthesis of Amines
46:05Synthesis of Amines
46:06
Gabriel Synthesis of Amines
47:57Gabriel Synthesis of Amines
47:58
Amines by SN2 with Azide Nu:
49:50Amines by SN2 with Azide Nu:
49:51
Amines by SN2 with Cyanide Nu:
50:31Amines by SN2 with Cyanide Nu:
50:32
Amines by Reduction of Amides
51:30Amines by Reduction of Amides
51:31
Reductive Amination of Ketones/Aldehydes
52:42Reductive Amination of Ketones/Aldehydes
52:43
Example : Synthesis of an Amine
53:47Example 1: Synthesis of an Amine
53:48Example 2: Synthesis of an Amine
56:16
Synthesis of Alkenes
58:20Synthesis of Alkenes Overview
58:21
Synthesis of Alkenes by Elimination
59:04Synthesis of Alkenes by Elimination Using NaOH & Heat
59:05Synthesis of Alkenes by Elimination Using H₂SO₄ & Heat
59:57
Synthesis of Alkenes by Reduction
1:02:05Alkyne to Cis Alkene
1:02:06Alkyne to Trans Alkene
1:02:56
Synthesis of Alkenes by Wittig Reaction
1:03:46Synthesis of Alkenes by Wittig Reaction
1:03:47Retrosynthesis of an Alkene
1:05:35
Example: Synthesis of an Alkene
1:06:57Example: Synthesis of an Alkene
1:06:58Making a Wittig Reagent
1:10:31
Synthesis of Alkynes
1:13:09Synthesis of Alkynes
1:13:10
Synthesis of Alkynes by Elimination (FGI)
1:13:42First Step: Bromination of Alkene
1:13:43Second Step: KOH Heat
1:14:22
Synthesis of Alkynes by Alkylation
1:15:02Synthesis of Alkynes by Alkylation
1:15:03Retrosynthesis of an Alkyne
1:16:18
Example: Synthesis of an Alkyne
1:17:40Example: Synthesis of an Alkyne
1:17:41
Synthesis of Alkanes
1:20:52Synthesis of Alkanes
1:20:53
Synthesis of Aldehydes & Ketones
1:21:38Oxidation of Alcohol Using PCC or Swern
1:21:39Oxidation of Alkene Using 1) O₃, 2)Zn
1:22:42Reduction of Acid Chloride & Nitrile Using DiBAL-H
1:23:25Hydration of Alkynes
1:24:55Synthesis of Ketones by Acyl Substitution
1:26:12Reaction with R'₂CuLi
1:26:13Reaction with R'MgBr
1:27:13
Synthesis of Aldehydes & Ketones by α-Alkylation
1:28:00Synthesis of Aldehydes & Ketones by α-Alkylation
1:28:01Retrosynthesis of a Ketone
1:30:10
Acetoacetate Ester Synthesis of Ketones
1:31:05Acetoacetate Ester Synthesis of Ketones: Step 1
1:31:06Acetoacetate Ester Synthesis of Ketones: Step 2
1:32:13Acetoacetate Ester Synthesis of Ketones: Step 3
1:32:50
Example: Synthesis of a Ketone
1:34:11Example: Synthesis of a Ketone
1:34:12
Synthesis of Carboxylic Acids
1:37:15Synthesis of Carboxylic Acids
1:37:16
Example: Synthesis of a Carboxylic Acid
1:37:59Example: Synthesis of a Carboxylic Acid (Option 1)
1:38:00Example: Synthesis of a Carboxylic Acid (Option 2)
1:40:51
Malonic Ester Synthesis of Carboxylic Acid
1:42:34Malonic Ester Synthesis of Carboxylic Acid: Step 1
1:42:35Malonic Ester Synthesis of Carboxylic Acid: Step 2
1:43:36Malonic Ester Synthesis of Carboxylic Acid: Step 3
1:44:01
Example: Synthesis of a Carboxylic Acid
1:44:53Example: Synthesis of a Carboxylic Acid
1:44:54
Synthesis of Carboxylic Acid Derivatives
1:48:05Synthesis of Carboxylic Acid Derivatives
1:48:06
Alternate Ester Synthesis
1:48:58Using Fischer Esterification
1:48:59Using SN2 Reaction
1:50:18Using Diazomethane
1:50:56Using 1) LDA, 2) R'-X
1:52:15
Practice: Synthesis of an Alkyl Chloride
1:53:11Practice: Synthesis of an Alkyl Chloride
1:53:12
Patterns of Functional Groups in Target Molecules
1:59:53Recall: Aldol Reaction
1:59:54β-hydroxy Ketone Target Molecule
2:01:12α,β-unsaturated Ketone Target Molecule
2:02:20
Patterns of Functional Groups in Target Molecules
2:03:15Recall: Michael Reaction
2:03:16Retrosynthesis: 1,5-dicarbonyl Target Molecule
2:04:07
Patterns of Functional Groups in Target Molecules
2:06:38Recall: Claisen Condensation
2:06:39Retrosynthesis: β-ketoester Target Molecule
2:07:30
2-Group Target Molecule Summary
2:09:032-Group Target Molecule Summary
2:09:04
Example: Synthesis of Epoxy Ketone
2:11:19Synthesize the Following Target Molecule from Cyclohexanone: Part 1 - Retrosynthesis
2:11:20Synthesize the Following Target Molecule from Cyclohexanone: Part 2 - Synthesis
2:14:10
Example: Synthesis of a Diketone
2:16:57Synthesis of a Diketone: Step 1 - Retrosynthesis
2:16:58Synthesis of a Diketone: Step 2 - Synthesis
2:18:51










































 Answer Engine
Answer Engine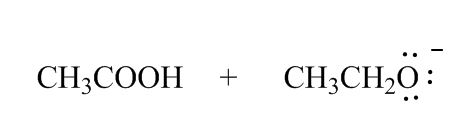
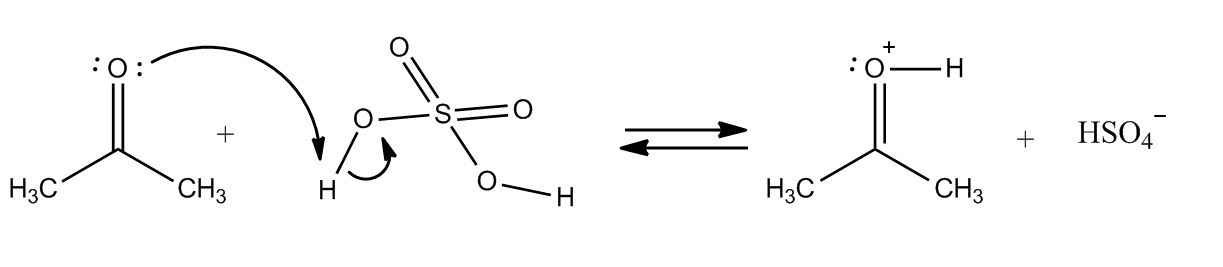
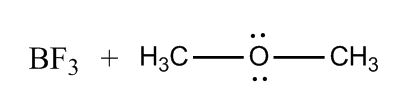
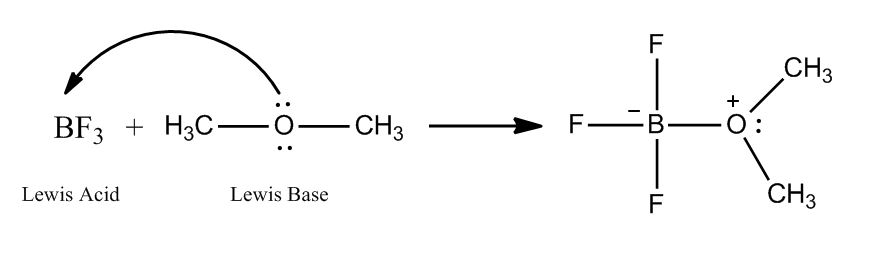








1 answer
Tue Aug 23, 2022 2:19 PM
Post by Mithil Krishnan on August 23, 2022
Hello,
I wanted to know what syllabus/textbook you are following, and what is the best websites to with practice problems/worksheets.
Please and thank you!
1 answer
Tue Mar 24, 2015 8:18 AM
Post by Sam Zanone on March 23, 2015
Good Morning Dr. Starkey,
I am currently enrolled in Advanced Organic Chemistry and using your amazing lectures to review the basic organic chemistry that I have not used for two years! Anyways, here is a question that I feel compelled to ask:
When discussing acid/base chemistry in general organic chemistry, we never mention the ultra-low and ultra-high pKa acids and bases (my professor calls them Super-Base's/crazy-weak acids). Why are these never brought up in basic organic chemistry, even briefly?
Thanks!
1 answer
Sat Feb 21, 2015 11:27 PM
Post by Saadman Elman on February 20, 2015
Hi Professor, Starkey. Hope you remembered me. It was a great lecture and it helped me a great deal. Thank you so much. I really don't have any question but i would like to make an interesting point. As i told you before that i signed up in educator.com 1 years ago and was taking general chemistry with Dr.OW. I figured that every professor has slightly different way to explain certain (gray)areas. I literally write down every single thing that u write and listen to your lecture before going to my class. You were explaining periodic trend for acidity (21:29) regarding which one is more acidic, HF, HCL, HBR, HI. You gave a very good justification why HI is the most acidic comparing to others and why HF is least acidic. My professor was going over the same topic in the class and asked us which one do we think is more acidic. I told him exactly what you said in this video. He said i made a lot of good points but he is not totally convinced. His logic regarding why HI is the most acidic is the following--- ''1)HI Has poor overlap, {As H has 1s orbital and I has 4p) 2) HI Has long bond, the longer the bond the easier it breaks and faster it ionizes than all others (HF,HCL,HBR) 3) Since, I- is larger so it can handle the (-) charge better, making it more delocalized.'' You made the last point in the video which he agrees.
1 answer
Tue Nov 11, 2014 12:13 AM
Post by Parth Shorey on November 10, 2014
Considering the factors that contribute to acidity, I don't understand how the middle one is not the most acidic. At 48:30 the inductive effect had the most stability but then again the aromatic compound had the resonance upper hand. How do I know which factor counts more?
1 answer
Sun Nov 9, 2014 10:09 PM
Post by Lalit Shorey on November 9, 2014
At 9:22, I don't understand how you determine what is acid or what is base besides using a Pka. Both show octet and lone pair. For some reason my prof won't give us a Pka table so what are other ways in this scenario?
1 answer
Fri Apr 25, 2014 12:18 AM
Post by Gina Weiland on April 24, 2014
Does anyone know any tips on figuring the pH ooh and oh of acid base reactions using logs?
2 answers
Last reply by: lakshmi tatineni
Sun Jan 5, 2014 12:30 PM
Post by lakshmi tatineni on January 2, 2014
I do not understand the importance of the lone pair on N and protanation?
1 answer
Sat Nov 30, 2013 1:39 AM
Post by John K on November 29, 2013
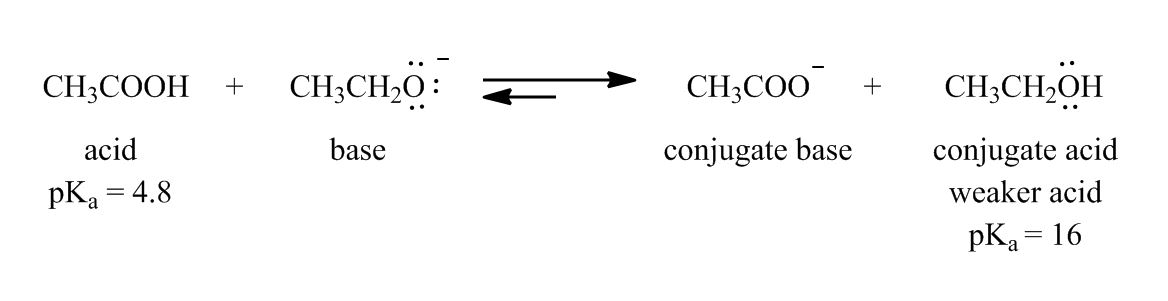
In the 3rd acid base example, why didn't we take conjugate acid to find out the strongest base?
1 answer
Sat Nov 30, 2013 1:42 AM
Post by John K on November 29, 2013
H20 is lewis base right? Then in Acid-Base equilibrium how did it become an acid?
thanks.
1 answer
Thu Nov 21, 2013 11:43 PM
Post by richa acharya on November 19, 2013
I'm not able to load any of the videos ever since I cancelled to extend the subscription however I thought I would be able to look at the videos till end of the month. I don't know how it works who should I talk to? I'm lost.
1 answer
Tue Oct 15, 2013 8:04 PM
Post by brandon oneal on October 15, 2013
Why didn't you add a plus charge to the oxygen for CH3CH2O(Resonance Effects Acidity)?
1 answer
Sun Sep 29, 2013 11:43 PM
Post by Ardeshir Badr on September 29, 2013
at around 3:00 why does AlCl3 only want 3 bonds and therefore with a - sign. and why does NH3 want 5 bonds and therefore has a + sign? please help!
1 answer
Sun Sep 15, 2013 9:13 PM
Post by Riley Argue on September 15, 2013
You are an excellent professor.
1 answer
Wed Sep 11, 2013 11:09 AM
Post by Frank Ofori-Addo on September 10, 2013
does the carbon that is found in the middle of the product of the second example have any formal charges? if no please state why. thanks.
1 answer
Sun Sep 8, 2013 1:19 PM
Post by Atreya Mohile on September 8, 2013
Is there any formula, that determines the number of resonating structures of a particular compound?
3 answers
Sun Sep 8, 2013 1:23 PM
Post by Atreya Mohile on September 5, 2013
As mentioned in lecture, that conjugate base of water, i.e. OH, is most stable, because O is more electro-ve, and has a higher capacity to hold -ve charge.. But if we apply the same rule to HX(X=halogens), their conj. bases give inverse of the real phenomena. So my question is that is this concept limited to periods of the periodic table only? Isn't it applicable to groups?
1 answer
Tue May 28, 2013 5:13 PM
Post by Tribhuwan Joshi on May 28, 2013
I know that this lecture isn't the correct place for this, but can you please tell me what the calorific value of methyl-propanol is?
Thanks a ton,
Piyush
1 answer
Fri May 10, 2013 8:42 AM
Post by Stephanie Bule on May 9, 2013
Professor Starky, on the energy diagram when you said that OH- was the least endothermic, what did you mean? I thought that low energy is more endothermic. I'm a little confused
Thank you!
1 answer
Sun Jan 27, 2013 12:30 AM
Post by Yao Mu on January 26, 2013
So if some compound has OH group that this compound can be acidic, as you mention in resonance effects on acidity, then why OH- always act as base.like NaOH?
1 answer
Sat Jan 26, 2013 9:43 PM
Post by marsha prytz on January 24, 2013
Dr Starky I was wondering how you come up with the energy table? I don't quite get how you know these molecules/CB have a particular level of energy.
0 answers
Post by Mori Jonata on October 27, 2012
you are right, i need to learn the name. Thanks
2 answers
Last reply by: Mori Jonata
Sat Oct 27, 2012 11:28 PM
Post by Mori Jonata on October 20, 2012
hello professor Starley. can u please explain how we get the negative charge on the nitrate ion (NO3-) and moreover, why do we have the same charge of the nitrite ion (NO2-). Thanks for you help
0 answers
Post by Mori Jonata on October 20, 2012
Thanks. appreciated
1 answer
Fri Oct 19, 2012 10:56 AM
Post by Mori Jonata on October 18, 2012
Dr Starkey, is educator.com for university student or just secondary school?
1 answer
Wed Oct 3, 2012 10:19 PM
Post by Mori Jonata on October 3, 2012
Thank you Dr starkey for the quick reply. I will recommend this website to a friend.
1 answer
Wed Oct 3, 2012 10:19 PM
Post by sophia lin on October 3, 2012
is that the resonance on the C.B always contribute the large amount to the stability?
1 answer
Tue Oct 2, 2012 11:30 PM
Post by Mori Jonata on October 1, 2012
How does the C in CHO pull electron toward itself. i thought O is more electronegative more than C and it should be pulling the electrons not the C. thanks
1 answer
Tue Oct 2, 2012 11:29 PM
Post by Mori Jonata on October 1, 2012
and moreover, which of the pull electrons toward theirself? is it the N in N02 or the O in N02. because i can see that the O in N02 has negative charge.
1 answer
Tue Oct 2, 2012 11:29 PM
Post by Mori Jonata on October 1, 2012
Hello professor Starkey, can you please explain the lewis structure behind the N02 of the topic (Inductive effect on Acidity). how do we get + charge on the nitrogendioxide(N02) and the one on cyni ion(CN)
1 answer
Tue Sep 18, 2012 10:44 AM
Post by Hawa Muse on September 15, 2012
how do you know which is a base and which is an acid?
1 answer
Thu Apr 12, 2012 11:31 PM
Post by Susan Barrett on April 11, 2012
I like how Professor Starkey used people as an example it really helped me remember and think of them as something more
2 answers
Sun Nov 25, 2012 12:15 AM
Post by Professor Starkey on April 10, 2012
Is there a way to skip to a certain section of the video?
1 answer
Sun Nov 20, 2011 9:19 AM
Post by WaiYee Hon on November 11, 2011
I m confused , so F is the most electronegative , why negative charge on F is not the most stable when comparing with Br- CI- or I- ?
3 answers
Mon Oct 15, 2012 9:49 PM
Post by Jindou Tian on October 11, 2011
Does this course only cover the material of the first semester of Organic Chemistry?
1 answer
Sun Sep 25, 2011 7:27 PM
Post by Bianca Williams on September 18, 2011
Is the size of the atom more important than the electronegativity of the atom when studying inductive effects? For example, if you had two molecules that deprotonated leaving I- and O- (and the rest of the molecule is the same for both), which of the two ions is going to have a stronger inductive effect?
1 answer
Fri Sep 9, 2011 11:34 PM
Post by Kangoma Kindembo on September 3, 2011
How can an element be known as the most electronegative by just looking at it? Does it depend on their emplacement in the periodic table?
1 answer
Wed Aug 17, 2011 3:44 PM
Post by Tej Jai on August 3, 2011
You said that CF3OH has a parent acid that is stronger (not Cf3Oh itself, but the parent acid). You also mention that CF3OH is a weaker conjugate base. So, how can it be a weaker conjugate base and a strong acid at the same time?
0 answers
Post by Professor Starkey on January 26, 2011
Awesome videos. Helpful in inductive effect.