Intro
0:00Organic Synthesis Strategies
0:15Goal
0:16Strategy
0:29
Example of a RetroSynthesis
1:30Finding Starting Materials for Target Molecule
1:31Synthesis Using Starting Materials
4:56
Synthesis of Alcohols by Functional Group Interconversion (FGI)
6:00Synthesis of Alcohols by Functional Group Interconversion Overview
6:01
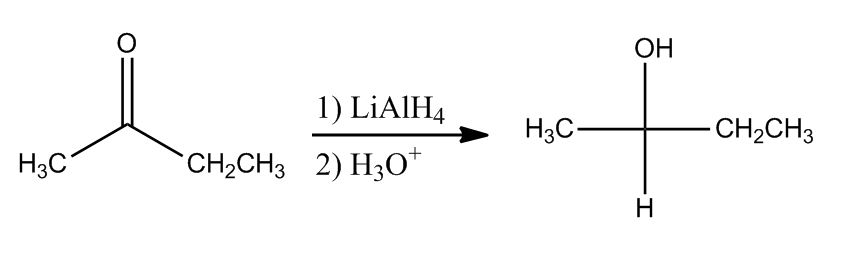
Alcohols by Reduction
7:43Ketone to Alcohols
7:45Aldehyde to Alcohols
8:26Carboxylic Acid Derivative to Alcohols
8:36
Alcohols by Hydration of Alkenes
9:28Hydration of Alkenes Using H₃O⁺
9:29Oxymercuration-Demercuration
10:35Hydroboration Oxidation
11:02
Alcohols by Substitution
11:42Primary Alkyl Halide to Alcohols Using NaOH
11:43Secondary Alkyl Halide to Alcohols Using Sodium Acetate
13:07Tertiary Alkyl Halide to Alcohols Using H₂O
15:08
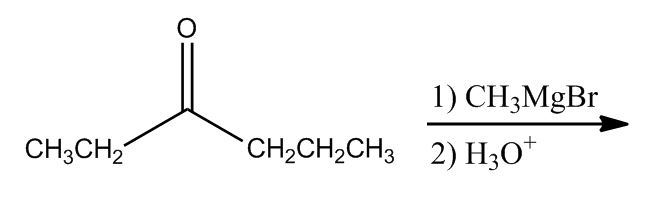
Synthesis of Alcohols by Forming a New C-C Bond
15:47Recall: Alcohol & RMgBr
15:48Retrosynthesis
17:28
Other Alcohol Disconnections
19:46
19:47Synthesis Using PhMGgBr: Example 2
23:05
Synthesis of Alkyl Halides
26:06Synthesis of Alkyl Halides Overview
26:07
Synthesis of Alkyl Halides by Free Radical Halogenation
27:04Synthesis of Alkyl Halides by Free Radical Halogenation
27:05
Synthesis of Alkyl Halides by Substitution
29:06Alcohol to Alkyl Halides Using HBr or HCl
29:07Alcohol to Alkyl Halides Using SOCl₂
30:57Alcohol to Alkyl Halides Using PBr₃ and Using P, I₂
31:03
Synthesis of Alkyl Halides by Addition
32:02Alkene to Alkyl Halides Using HBr
32:03Alkene to Alkyl Halides Using HBr & ROOR (Peroxides)
32:35
Example: Synthesis of Alkyl Halide
34:18Example: Synthesis of Alkyl Halide
34:19
Synthesis of Ethers
39:25Synthesis of Ethers
39:26
Example: Synthesis of an Ether
41:12Synthesize TBME (t-butyl methyl ether) from Alcohol Starting Materials
41:13
Synthesis of Amines
46:05Synthesis of Amines
46:06
Gabriel Synthesis of Amines
47:57Gabriel Synthesis of Amines
47:58
Amines by SN2 with Azide Nu:
49:50Amines by SN2 with Azide Nu:
49:51
Amines by SN2 with Cyanide Nu:
50:31Amines by SN2 with Cyanide Nu:
50:32
Amines by Reduction of Amides
51:30Amines by Reduction of Amides
51:31
Reductive Amination of Ketones/Aldehydes
52:42Reductive Amination of Ketones/Aldehydes
52:43
Example : Synthesis of an Amine
53:47Example 1: Synthesis of an Amine
53:48Example 2: Synthesis of an Amine
56:16
Synthesis of Alkenes
58:20Synthesis of Alkenes Overview
58:21
Synthesis of Alkenes by Elimination
59:04Synthesis of Alkenes by Elimination Using NaOH & Heat
59:05Synthesis of Alkenes by Elimination Using H₂SO₄ & Heat
59:57
Synthesis of Alkenes by Reduction
1:02:05Alkyne to Cis Alkene
1:02:06Alkyne to Trans Alkene
1:02:56
Synthesis of Alkenes by Wittig Reaction
1:03:46Synthesis of Alkenes by Wittig Reaction
1:03:47Retrosynthesis of an Alkene
1:05:35
Example: Synthesis of an Alkene
1:06:57Example: Synthesis of an Alkene
1:06:58Making a Wittig Reagent
1:10:31
Synthesis of Alkynes
1:13:09Synthesis of Alkynes
1:13:10
Synthesis of Alkynes by Elimination (FGI)
1:13:42First Step: Bromination of Alkene
1:13:43Second Step: KOH Heat
1:14:22
Synthesis of Alkynes by Alkylation
1:15:02Synthesis of Alkynes by Alkylation
1:15:03Retrosynthesis of an Alkyne
1:16:18
Example: Synthesis of an Alkyne
1:17:40Example: Synthesis of an Alkyne
1:17:41
Synthesis of Alkanes
1:20:52Synthesis of Alkanes
1:20:53
Synthesis of Aldehydes & Ketones
1:21:38Oxidation of Alcohol Using PCC or Swern
1:21:39Oxidation of Alkene Using 1) O₃, 2)Zn
1:22:42Reduction of Acid Chloride & Nitrile Using DiBAL-H
1:23:25Hydration of Alkynes
1:24:55Synthesis of Ketones by Acyl Substitution
1:26:12Reaction with R'₂CuLi
1:26:13Reaction with R'MgBr
1:27:13
Synthesis of Aldehydes & Ketones by α-Alkylation
1:28:00Synthesis of Aldehydes & Ketones by α-Alkylation
1:28:01Retrosynthesis of a Ketone
1:30:10
Acetoacetate Ester Synthesis of Ketones
1:31:05Acetoacetate Ester Synthesis of Ketones: Step 1
1:31:06Acetoacetate Ester Synthesis of Ketones: Step 2
1:32:13Acetoacetate Ester Synthesis of Ketones: Step 3
1:32:50
Example: Synthesis of a Ketone
1:34:11Example: Synthesis of a Ketone
1:34:12
Synthesis of Carboxylic Acids
1:37:15Synthesis of Carboxylic Acids
1:37:16
Example: Synthesis of a Carboxylic Acid
1:37:59Example: Synthesis of a Carboxylic Acid (Option 1)
1:38:00Example: Synthesis of a Carboxylic Acid (Option 2)
1:40:51
Malonic Ester Synthesis of Carboxylic Acid
1:42:34Malonic Ester Synthesis of Carboxylic Acid: Step 1
1:42:35Malonic Ester Synthesis of Carboxylic Acid: Step 2
1:43:36Malonic Ester Synthesis of Carboxylic Acid: Step 3
1:44:01
Example: Synthesis of a Carboxylic Acid
1:44:53Example: Synthesis of a Carboxylic Acid
1:44:54
Synthesis of Carboxylic Acid Derivatives
1:48:05Synthesis of Carboxylic Acid Derivatives
1:48:06
Alternate Ester Synthesis
1:48:58Using Fischer Esterification
1:48:59Using SN2 Reaction
1:50:18Using Diazomethane
1:50:56Using 1) LDA, 2) R'-X
1:52:15
Practice: Synthesis of an Alkyl Chloride
1:53:11Practice: Synthesis of an Alkyl Chloride
1:53:12
Patterns of Functional Groups in Target Molecules
1:59:53Recall: Aldol Reaction
1:59:54β-hydroxy Ketone Target Molecule
2:01:12α,β-unsaturated Ketone Target Molecule
2:02:20
Patterns of Functional Groups in Target Molecules
2:03:15Recall: Michael Reaction
2:03:16Retrosynthesis: 1,5-dicarbonyl Target Molecule
2:04:07
Patterns of Functional Groups in Target Molecules
2:06:38Recall: Claisen Condensation
2:06:39Retrosynthesis: β-ketoester Target Molecule
2:07:30
2-Group Target Molecule Summary
2:09:032-Group Target Molecule Summary
2:09:04
Example: Synthesis of Epoxy Ketone
2:11:19Synthesize the Following Target Molecule from Cyclohexanone: Part 1 - Retrosynthesis
2:11:20Synthesize the Following Target Molecule from Cyclohexanone: Part 2 - Synthesis
2:14:10
Example: Synthesis of a Diketone
2:16:57Synthesis of a Diketone: Step 1 - Retrosynthesis
2:16:58Synthesis of a Diketone: Step 2 - Synthesis
2:18:51










































 Answer Engine
Answer Engine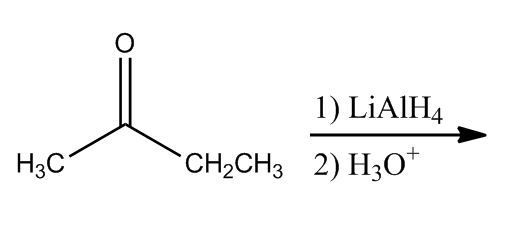
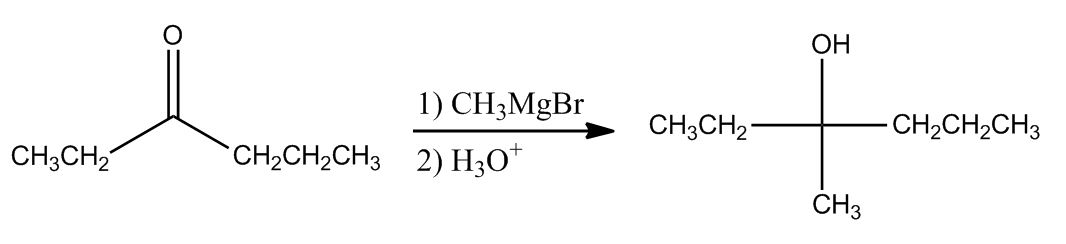
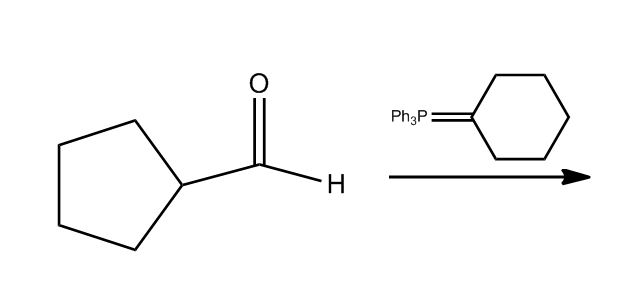
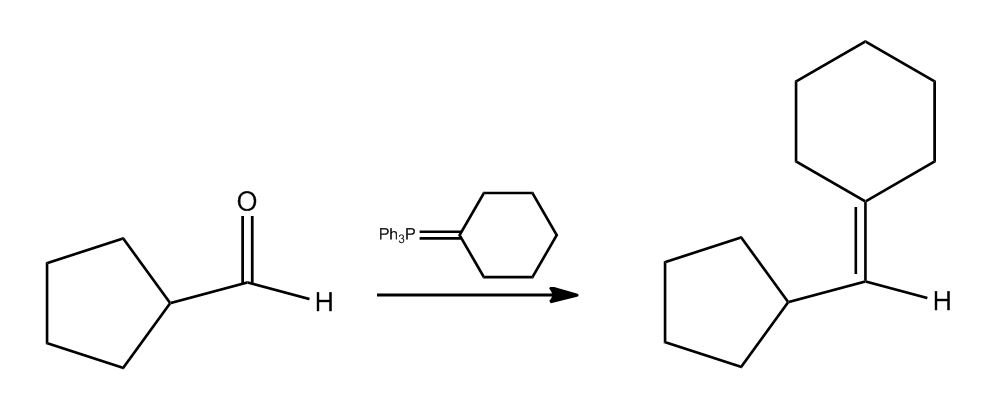
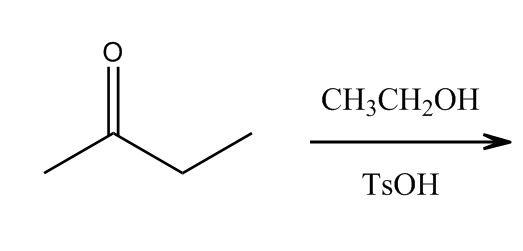
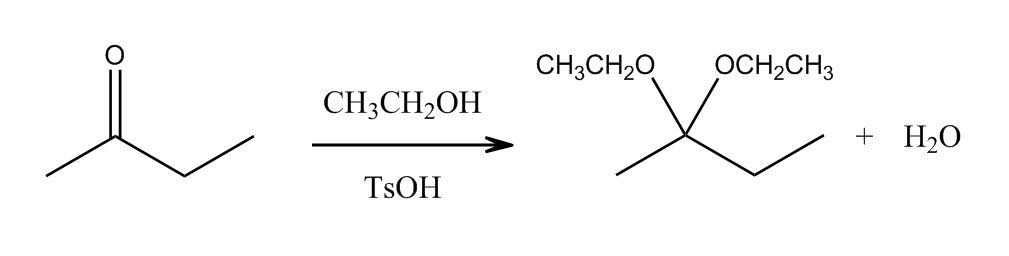
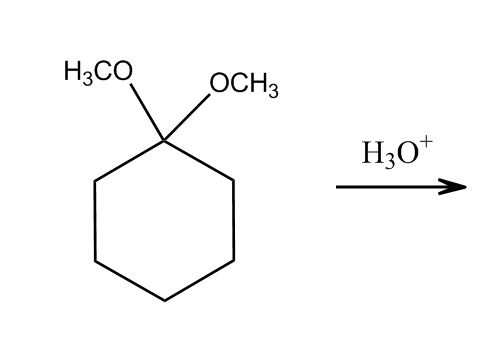
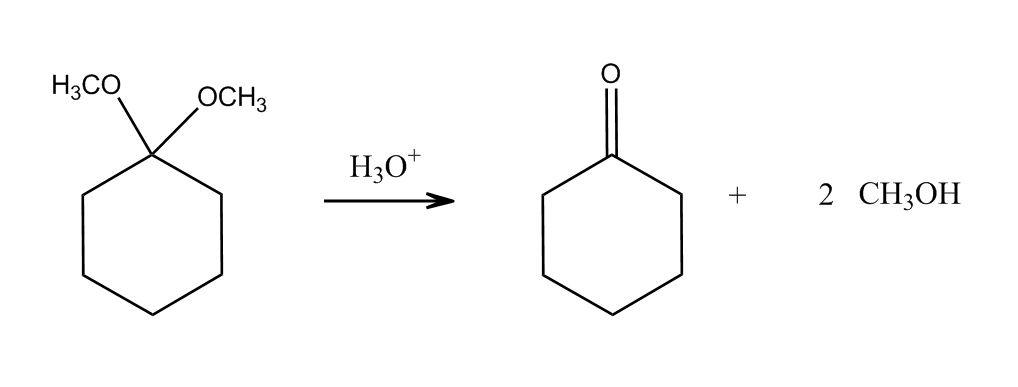
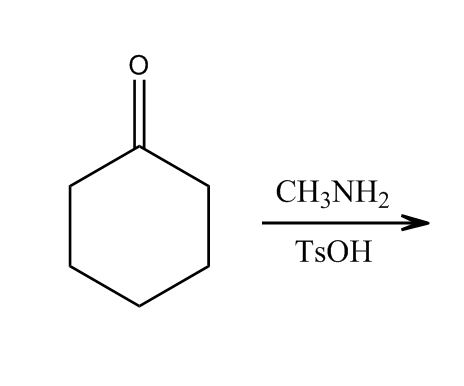







1 answer
Tue Jul 13, 2021 6:03 PM
Post by jacksonhogenson on May 17, 2021
At approx. 33 min. into the video, where you synthesize butanol from propanol, you first use PBR3 to get propyl bromide. You then use Mg to get a Grignard reagent. After which, you use (1) epoxide and then (2) H3O+ to get the final product. My question is: How do you know whether to put the OH on the more or less substituted carbon? I thought OH would have been on the more substituted carbon (i.e., in one from the end). Thanks!
1 answer
Mon Apr 4, 2016 2:12 PM
Post by Jordan Arciniega on April 3, 2016
Hello Dr. Starkey,
During the acetal mechanism, the first step is protonation of the carbonyl oxygen. This results in the formal charge of the oxygen to be plus one.
But why exactly does this result in the carbon of the carbonyl group to be more electrophilic? Is it because the plus charge on the oxygen changes its electronegativity such that it pulls electron density even more to itself (inductive effect), leaving the carbon more positively charged? Or is it because the oxygen with the positive charge now favors the resonance form of a plus charge on the carbon (to make oxygen more happy)?
I guess in general, I would like to know if plus charges on atoms change the electronegativity of that atom. So if an atom becomes a plus 1 charge, will it attract more electron density towards itself compared to its neutral form?
Thanks in advance, and like always, I highly appreciate these extremely helpful lectures!
1 answer
Wed Mar 30, 2016 3:15 PM
Post by Jonathan Ibera on March 29, 2016
Thanks, your lecture are very clear.
For the clemmensen reduction of ketones and aldehydes all the way to alkane, can this reduction method reduce alcohol as well? I know this is kind of gray area as the mechanism of clemmensen reduction might not be clear to me.
1 answer
Thu Feb 25, 2016 12:46 PM
Post by Milki Hussen on February 25, 2016
Is it possible to use to use methanal + a 4 carbon grignard reagent to get the propanol on on retrosynthesis ( 28 minutes)?
1 answer
Tue Jan 26, 2016 9:27 PM
Post by Jason Smith on January 26, 2016
Hi professor. In the formation of acetals, the reason why the acid acts first is because it's faster? Thank you.
1 answer
Sun Dec 14, 2014 8:31 PM
Post by bea v on December 14, 2014
if an C=O bond is flaked by an oxygen on both side is it still more reactive than a C=C ?
1 answer
Wed Mar 19, 2014 9:13 PM
Post by Jude Nawlo on March 19, 2014
Hi Dr. Starkey,
Do the same concepts that apply in Wittig apply for Horner-Wadsworth Emmons reactions?
3 answers
Mon Mar 3, 2014 10:23 PM
Post by Florel Fraser on March 1, 2014
I am having problems with the portion of the video on Wittig Reagent. As soon as Dr. Starkey starts the mechanism the video stops and goes back to the beginning of the lesson.
2 answers
Wed Jul 19, 2017 11:01 AM
Post by Victor Ye on May 27, 2013
I've really enjoyed your lessons, Prof. Starkey!! They're VERY helpful, and they're the best chemistry video lectures I've found!
I also second Nawaphan's comment about possibly covering biological molecules in your lessons. Educator has a biochemistry section, but I think lessons with a chemistry perspective would very useful for us. My OChem class covers these biological molecules, and the last three chapters of my textbook concern these topics. Thank you!
2 answers
Wed Jul 19, 2017 10:59 AM
Post by Nawaphan Jedjomnongkit on May 3, 2013
You are great teacher and this is the first time that I enjoy every ORG Chem lesson!! Now I can remember CTI because it's always come with sound ding ding ding!!!!! lol Thank you so much!! Is it possible to add some lessons about biological molecules like proteins, amino acids, carbohydrates and lipids?
1 answer
Sun May 20, 2012 10:13 AM
Post by Mark Deming on May 17, 2012
Wouldn't the grignard add twice so you should use lithium for first addition. That way you won't get two isopropyl groups?
0 answers
Post by Jason Jarduck on March 3, 2012
Hi Dr. Starkey,
Another great lecture. I have been taking notes and using your lectures for studying for tests and exams.
Thank You
Jason
1 answer
Wed Nov 30, 2011 11:13 PM
Post by Professor Starkey on November 30, 2011
Thank you so much Dr Starkey, the best OCHM teacher ever!!!
2 answers
Sat Nov 5, 2011 3:15 PM
Post by Clint Rapp on October 29, 2011
this is a Ketal because a ketone was the starting material? What is up with that?
1 answer
Sat Oct 8, 2011 3:01 PM
Post by Gayk Gevorkyan on October 5, 2011
Wouldn't be able to PASS OCHEM without this......THANK YOU DR.LAURIE!