Intro
0:00Organic Synthesis Strategies
0:15Goal
0:16Strategy
0:29
Example of a RetroSynthesis
1:30Finding Starting Materials for Target Molecule
1:31Synthesis Using Starting Materials
4:56
Synthesis of Alcohols by Functional Group Interconversion (FGI)
6:00Synthesis of Alcohols by Functional Group Interconversion Overview
6:01
Alcohols by Reduction
7:43Ketone to Alcohols
7:45Aldehyde to Alcohols
8:26Carboxylic Acid Derivative to Alcohols
8:36
Alcohols by Hydration of Alkenes
9:28Hydration of Alkenes Using H₃O⁺
9:29Oxymercuration-Demercuration
10:35Hydroboration Oxidation
11:02
Alcohols by Substitution
11:42Primary Alkyl Halide to Alcohols Using NaOH
11:43Secondary Alkyl Halide to Alcohols Using Sodium Acetate
13:07Tertiary Alkyl Halide to Alcohols Using H₂O
15:08
Synthesis of Alcohols by Forming a New C-C Bond
15:47Recall: Alcohol & RMgBr
15:48Retrosynthesis
17:28
Other Alcohol Disconnections
19:46
19:47Synthesis Using PhMGgBr: Example 2
23:05
Synthesis of Alkyl Halides
26:06Synthesis of Alkyl Halides Overview
26:07
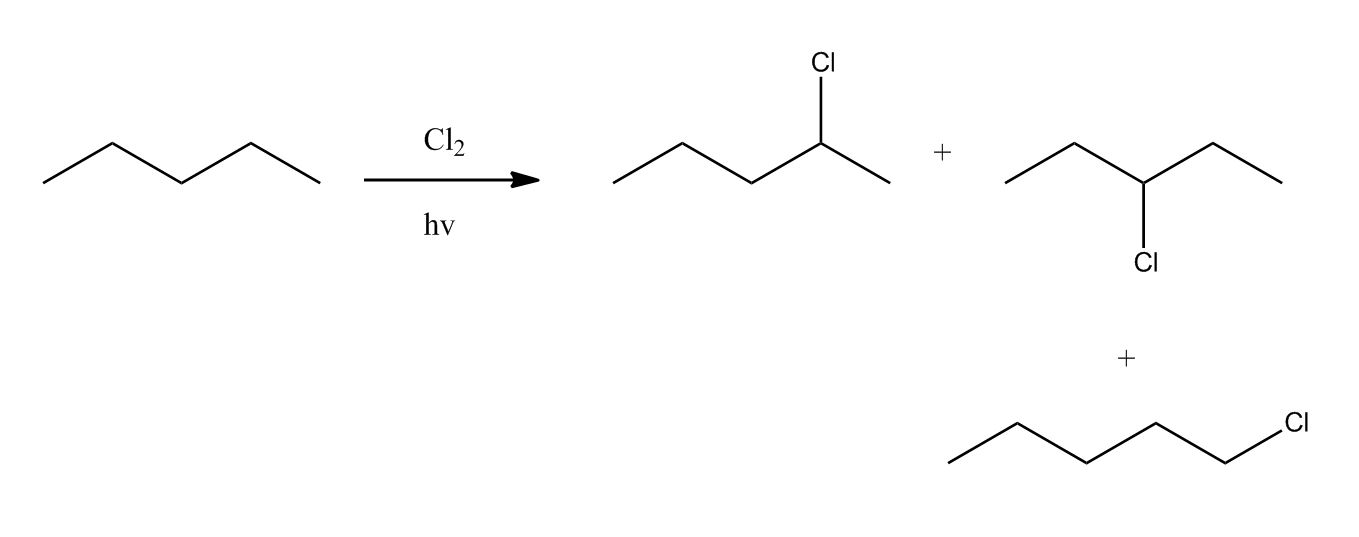
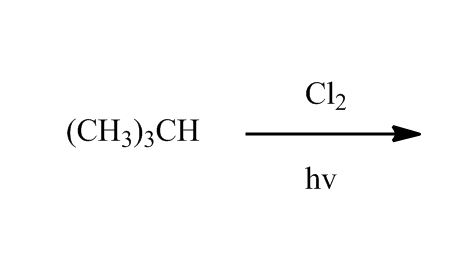
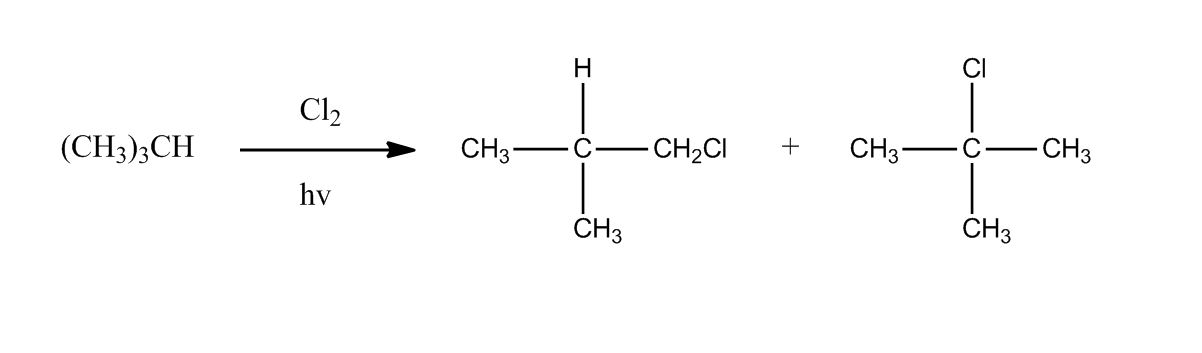
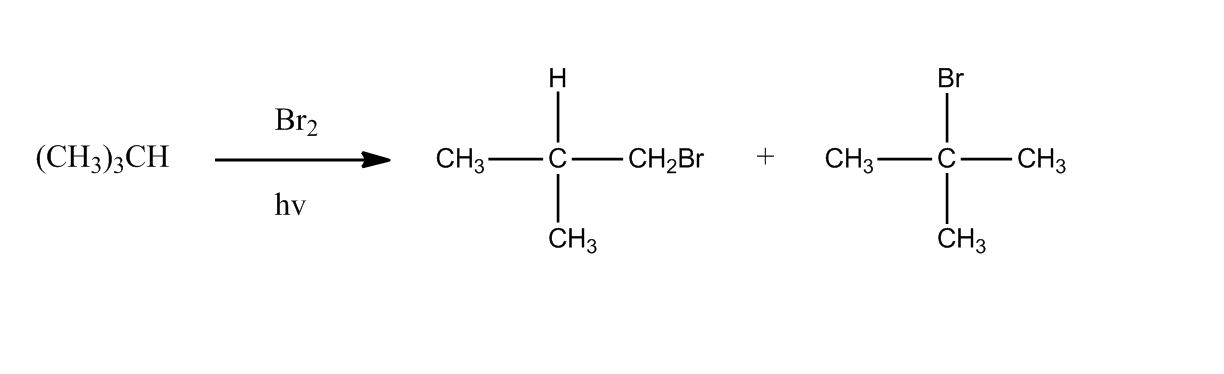
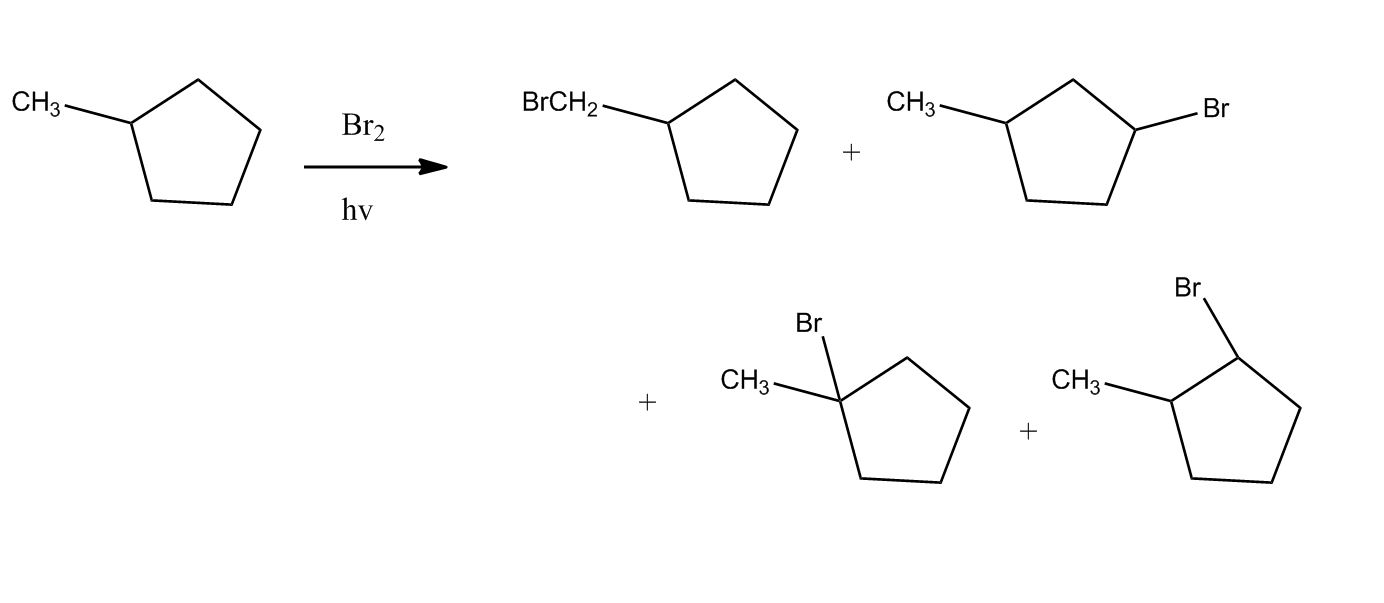
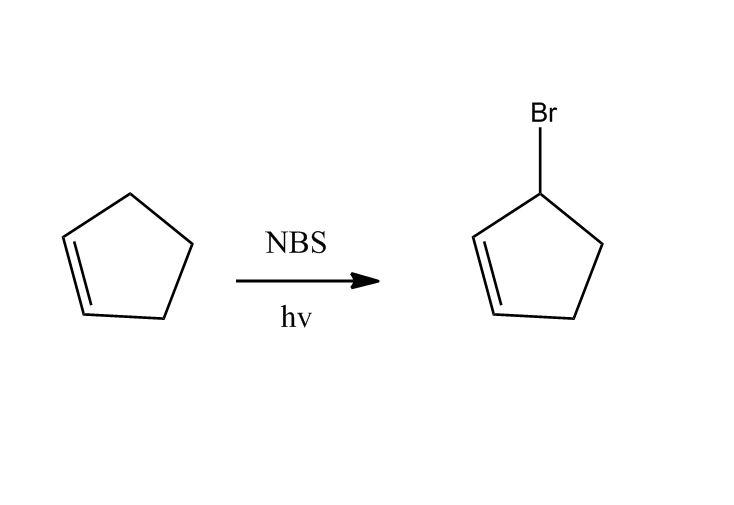
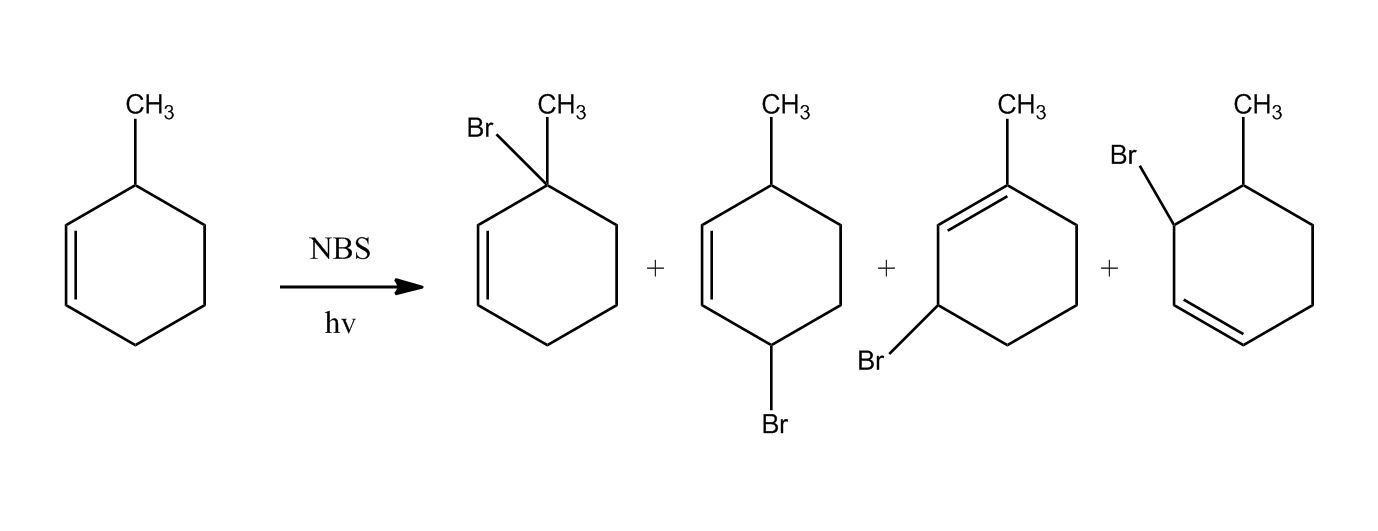
Synthesis of Alkyl Halides by Free Radical Halogenation
27:04Synthesis of Alkyl Halides by Free Radical Halogenation
27:05
Synthesis of Alkyl Halides by Substitution
29:06Alcohol to Alkyl Halides Using HBr or HCl
29:07Alcohol to Alkyl Halides Using SOCl₂
30:57Alcohol to Alkyl Halides Using PBr₃ and Using P, I₂
31:03
Synthesis of Alkyl Halides by Addition
32:02Alkene to Alkyl Halides Using HBr
32:03Alkene to Alkyl Halides Using HBr & ROOR (Peroxides)
32:35
Example: Synthesis of Alkyl Halide
34:18Example: Synthesis of Alkyl Halide
34:19
Synthesis of Ethers
39:25Synthesis of Ethers
39:26
Example: Synthesis of an Ether
41:12Synthesize TBME (t-butyl methyl ether) from Alcohol Starting Materials
41:13
Synthesis of Amines
46:05Synthesis of Amines
46:06
Gabriel Synthesis of Amines
47:57Gabriel Synthesis of Amines
47:58
Amines by SN2 with Azide Nu:
49:50Amines by SN2 with Azide Nu:
49:51
Amines by SN2 with Cyanide Nu:
50:31Amines by SN2 with Cyanide Nu:
50:32
Amines by Reduction of Amides
51:30Amines by Reduction of Amides
51:31
Reductive Amination of Ketones/Aldehydes
52:42Reductive Amination of Ketones/Aldehydes
52:43
Example : Synthesis of an Amine
53:47Example 1: Synthesis of an Amine
53:48Example 2: Synthesis of an Amine
56:16
Synthesis of Alkenes
58:20Synthesis of Alkenes Overview
58:21
Synthesis of Alkenes by Elimination
59:04Synthesis of Alkenes by Elimination Using NaOH & Heat
59:05Synthesis of Alkenes by Elimination Using H₂SO₄ & Heat
59:57
Synthesis of Alkenes by Reduction
1:02:05Alkyne to Cis Alkene
1:02:06Alkyne to Trans Alkene
1:02:56
Synthesis of Alkenes by Wittig Reaction
1:03:46Synthesis of Alkenes by Wittig Reaction
1:03:47Retrosynthesis of an Alkene
1:05:35
Example: Synthesis of an Alkene
1:06:57Example: Synthesis of an Alkene
1:06:58Making a Wittig Reagent
1:10:31
Synthesis of Alkynes
1:13:09Synthesis of Alkynes
1:13:10
Synthesis of Alkynes by Elimination (FGI)
1:13:42First Step: Bromination of Alkene
1:13:43Second Step: KOH Heat
1:14:22
Synthesis of Alkynes by Alkylation
1:15:02Synthesis of Alkynes by Alkylation
1:15:03Retrosynthesis of an Alkyne
1:16:18
Example: Synthesis of an Alkyne
1:17:40Example: Synthesis of an Alkyne
1:17:41
Synthesis of Alkanes
1:20:52Synthesis of Alkanes
1:20:53
Synthesis of Aldehydes & Ketones
1:21:38Oxidation of Alcohol Using PCC or Swern
1:21:39Oxidation of Alkene Using 1) O₃, 2)Zn
1:22:42Reduction of Acid Chloride & Nitrile Using DiBAL-H
1:23:25Hydration of Alkynes
1:24:55Synthesis of Ketones by Acyl Substitution
1:26:12Reaction with R'₂CuLi
1:26:13Reaction with R'MgBr
1:27:13
Synthesis of Aldehydes & Ketones by α-Alkylation
1:28:00Synthesis of Aldehydes & Ketones by α-Alkylation
1:28:01Retrosynthesis of a Ketone
1:30:10
Acetoacetate Ester Synthesis of Ketones
1:31:05Acetoacetate Ester Synthesis of Ketones: Step 1
1:31:06Acetoacetate Ester Synthesis of Ketones: Step 2
1:32:13Acetoacetate Ester Synthesis of Ketones: Step 3
1:32:50
Example: Synthesis of a Ketone
1:34:11Example: Synthesis of a Ketone
1:34:12
Synthesis of Carboxylic Acids
1:37:15Synthesis of Carboxylic Acids
1:37:16
Example: Synthesis of a Carboxylic Acid
1:37:59Example: Synthesis of a Carboxylic Acid (Option 1)
1:38:00Example: Synthesis of a Carboxylic Acid (Option 2)
1:40:51
Malonic Ester Synthesis of Carboxylic Acid
1:42:34Malonic Ester Synthesis of Carboxylic Acid: Step 1
1:42:35Malonic Ester Synthesis of Carboxylic Acid: Step 2
1:43:36Malonic Ester Synthesis of Carboxylic Acid: Step 3
1:44:01
Example: Synthesis of a Carboxylic Acid
1:44:53Example: Synthesis of a Carboxylic Acid
1:44:54
Synthesis of Carboxylic Acid Derivatives
1:48:05Synthesis of Carboxylic Acid Derivatives
1:48:06
Alternate Ester Synthesis
1:48:58Using Fischer Esterification
1:48:59Using SN2 Reaction
1:50:18Using Diazomethane
1:50:56Using 1) LDA, 2) R'-X
1:52:15
Practice: Synthesis of an Alkyl Chloride
1:53:11Practice: Synthesis of an Alkyl Chloride
1:53:12
Patterns of Functional Groups in Target Molecules
1:59:53Recall: Aldol Reaction
1:59:54β-hydroxy Ketone Target Molecule
2:01:12α,β-unsaturated Ketone Target Molecule
2:02:20
Patterns of Functional Groups in Target Molecules
2:03:15Recall: Michael Reaction
2:03:16Retrosynthesis: 1,5-dicarbonyl Target Molecule
2:04:07
Patterns of Functional Groups in Target Molecules
2:06:38Recall: Claisen Condensation
2:06:39Retrosynthesis: β-ketoester Target Molecule
2:07:30
2-Group Target Molecule Summary
2:09:032-Group Target Molecule Summary
2:09:04
Example: Synthesis of Epoxy Ketone
2:11:19Synthesize the Following Target Molecule from Cyclohexanone: Part 1 - Retrosynthesis
2:11:20Synthesize the Following Target Molecule from Cyclohexanone: Part 2 - Synthesis
2:14:10
Example: Synthesis of a Diketone
2:16:57Synthesis of a Diketone: Step 1 - Retrosynthesis
2:16:58Synthesis of a Diketone: Step 2 - Synthesis
2:18:51










































 Answer Engine
Answer Engine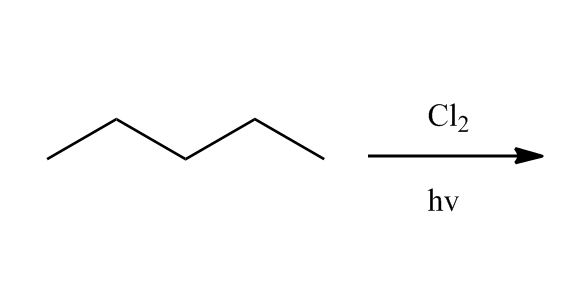








1 answer
Mon Feb 18, 2019 11:36 PM
Post by Curtis Marriott on February 17, 2019
professor, your videos are great,but at the 1:20 :39 mark it keeps looping. this has occurred with some of the other videos as well
1 answer
Wed Aug 8, 2018 9:31 AM
Post by Mohammad Abdel-halim on July 24, 2018
Hello Professor,
Example 4 at min 66, will the product coming from the allylic carbocation which has the charge on the 2ry carbon the major one?
Thank you
1 answer
Sat Jan 13, 2018 7:30 PM
Post by Ofonime Emah on December 2, 2017
Hello, is there solution steps or something explaining how the product/answer was derived for the practice questions?
2 answers
Last reply by: Matthew Zhang
Wed Jul 19, 2017 6:45 PM
Post by Avery Dawes on July 19, 2017
Does anyone know why I might not be able to fast forward? I have already watched half the lecture and I would like to pick up where I left off but I can't seem to figure out how to do so.
2 answers
Last reply by: Jinbin Chen
Sun Jul 26, 2015 3:55 PM
Post by Jinbin Chen on July 25, 2015
Hi, Dr. Starkey!
So for tertiary substrates with OH as the leaving group, we need to protonate the OH to convert it to HOH. But if we are given both a strong acid and a polar protic solvent (like EtOH, which also functions as the nucleophile), which species will actually protonate the LG, the strong acid (H3O+) or the protonated solvent molecule (EtOH2+)?
Also, if we are asked to provide reagents for Sn2 reactions, is it necessary to write down the solvent used alongside the nucleophiles (such as NaI/DMSO)? The solution manual I used does not always include the solvent as part of the answer.
1 answer
Wed Dec 10, 2014 10:30 PM
Post by Camille Fraser on December 9, 2014
NO VOLUME...FOR THE SECOND TIME..
ON Sunday the feed kept on looping for at least an hour..FRUSTRATING!!
THE sad part is that I REALLY Don't have time for this..
1 answer
Mon Nov 17, 2014 10:41 PM
Post by Parth Shorey on November 16, 2014
At 67:01 I don't understand the third problem, what about the backside attack? Doesn't it block it ?
1 answer
Sun Nov 16, 2014 12:31 PM
Post by Parth Shorey on November 15, 2014
What happened to the Cl? At 38:45 I am still confused because Cl is more electronegative and is a good leaving group but you used fast reaction? I still don't understand why that happened?
1 answer
Sat Nov 1, 2014 11:28 PM
Post by Steven Pulido on October 30, 2014
you said allec compounds do sn2 fast and next say the same to sn1! doesnt make sense!
2 answers
Last reply by: David Gonzalez
Fri Oct 31, 2014 5:18 PM
Post by David Gonzalez on October 28, 2014
Hello Professor Starkey, I have two questions that are really baffling me. First, why is Fluorine considered a "poor" leaving group? Also, why are tertiary carbocations considered more stable than primary carbocations? Thanks!
3 answers
Sun Nov 2, 2014 1:47 PM
Post by Rene Whitaker on October 26, 2014
I have tried playing this lecture several times and the quality is terrible. The sound keeps skipping. I am having no other computer trouble and this is the first one here that does this...any suggestions?
1 answer
Fri Apr 25, 2014 12:22 AM
Post by Saria Abbas on April 24, 2014
Where can I find the practice questions for organic chemistry?
1 answer
Mon Mar 24, 2014 5:18 PM
Post by minal patel on March 23, 2014
I am trying to look for an appropriate lecture for benzene substitution reactions. and the otho meta and para effects
1 answer
Wed Jan 29, 2014 10:41 PM
Post by Yanet Ortiz on January 28, 2014
good morning professor:
I have a question at 18:21. You said it is an endothermic rxn, I thought it is an exothermic reaction. Can you explain that part for me please? Thank you for your lectures. They have been very helpful for me!!!
1 answer
Last reply by: Jeffrey Zhang
Sun Sep 1, 2013 8:23 PM
Post by Jeffrey Zhang on September 1, 2013
Hi Dr. Starkey,
Would it be possible to introduce TsI for the synthesis problem at 98:15 so that we would not have to add an additional solution of NaI?
Thanks!
Jeff Z
1 answer
Tue Aug 13, 2013 11:41 AM
Post by Briana Kallias on August 11, 2013
At 54:58, where would the plane of symmetry be drawn on the planar molecule? I understand it can't be chiral because it doesn't have 4 separate groups on the carbon, but I can't see the plane of symmetry you referred to.
Thanks
1 answer
Wed Jun 5, 2013 10:43 PM
Post by Kate Bevan on June 5, 2013
These lectures are so helpful. It's amazing how much easier good teaching can make things! Thank you!
1 answer
Mon May 27, 2013 2:05 PM
Post by Morla Lochhead on May 27, 2013
In the monofluoridation of a multi branched alkane, I would assume it to be a SN1 reaction...there is no leaving group in the form of a Halogen but the primary carbons and methyl groups are the most vulnerable leaving a carbocation as the electrophile and the floride has a opportunity to bond at that sight. Am I going in the right direction? Any additional resources that you recommend?
1 answer
Sun Apr 28, 2013 11:10 PM
Post by Abraham Rouzyi on April 28, 2013
At 54:29 isnt it the other way around, the R is inversion and the S retention because of how the methyl groups are facing in the same direction as they do in the intermediate. Can you explain that please? thank you.
1 answer
Fri Apr 26, 2013 12:53 AM
Post by Alicia DaSilva on April 25, 2013
Good day Professor Starkey,
when explaining stereochemistry for both the SN2 and SN1 examples you used a chiral carbon that has a methyl, H, Cl and ethyl group attached, how would I determine which mechanism to use, would the indicator be the type of Nu:, it is just a little confusing; since, the same compound was used in both example.
1 answer
Tue Apr 9, 2013 12:13 AM
Post by Conor Brady on April 8, 2013
Hi Doctor Starkey
Sorry but how do you tell if a base is strong or weak to determine what reaction it will go through? Do you use a pka table?
Thanks Conor
1 answer
Sun Jan 6, 2013 1:25 PM
Post by Aaron Harper on January 6, 2013
For 80:26, Professor Starkey finishes the mechanism reaction, but she deprotinated with H2O, when she meant it would be Br. So in other words, our products were the alcohol she mentioned and HBr, not H3O.
Just FYI. Other than that, Professor Starkey has done phenomenally to this point. I wish I'd have had her when I took Organics. Now, because of Educator, and these lectures, I've been able to review Organics during winter break. AND I actually understand what's going on :)
Thank you all!
2 answers
Sun Dec 16, 2012 4:56 PM
Post by natasha plantak on December 16, 2012
In 65:05 isn't CH3COOH a weak nucleophile, so wouldn't this reaction favor Sn1?
2 answers
Last reply by: natasha plantak
Mon Dec 17, 2012 6:53 AM
Post by natasha plantak on December 16, 2012
In 64;06 I dont see how b is an achiral molecule (mess compound). Since one side has a methyl and the other is adding an I, doesn't that make the molecule chiral. There is no plane of symmetry since CH3 and I are not the same?
1 answer
Thu Nov 15, 2012 12:52 AM
Post by Anthony Blair on November 14, 2012
Addition reactions in your lectures somewhere??
2 answers
Last reply by: Anthony Blair
Fri Nov 16, 2012 10:02 AM
Post by Anthony Blair on November 13, 2012
Hello,
Under Organic Chemistry, I see substitution reactions and elimination reactions. However, my next test next Tuesday is over the content involving substitution reactions, elimination reactions, addition reactions. Where under Organic Chemistry is addition reactions content? Is addition reactions under another heading? I need to understand this material for my next test. I somehow need to do really well on my next test. My schools organic professors just points at the powerpoint. I need someone to explain the stuff. I am going back to the college in the spring for organic chemistry two college where I did my gen chems because they are retired chemical engineers and are awesome explaining chemistry and teach from their own notes and know the material and do not use power points.
Anyways, where do I find addition reactions within the organic chemistry section? It should be here somewhere if substitution and elimination is on here. I noticed during my last subscription that mass spec was not on here, and luckily I finally understood on my own the chapter enough to do well in that section on the last test.
Thank you
1 answer
Mon Nov 12, 2012 10:36 PM
Post by Paula Hoggard on November 12, 2012
You're an amazing teacher. If ever I miss a class I come watch your videos. You make organic chemistry so simple.
Thank you so much
Paula
1 answer
Wed Oct 31, 2012 9:18 PM
Post by Melissa Mondo on October 31, 2012
I saw someone else asked this question as well but I can't get the answers to open;
When I see a reaction with one molecule on top of the reaction arrow and one below the reaction arrow. Is that telling me that the top is the Nu: and the bottom is the solvent?
I had a problem with an alkyl bromide carried out two different ways, one with CH3OH over the arrow, and one with CH3O- above the arrow with DMSO below the arrow.
I understood in my 1st case but with the 2nd case I don't understand why a solvent is needed shouldn't the Br be a good LG without the solvent?
1 answer
Fri Oct 26, 2012 12:40 AM
Post by Kelly Corona on October 24, 2012
Can you explain the chemistry behind: A) 41:20- why does the 3 carbon ring form? If is high strain, doesn’t that mean is very unstable?
1 answer
Wed Oct 24, 2012 7:56 PM
Post by Kelly Corona on October 24, 2012
Can you discuss the specifics of why p-orbitals are better? I know you said because of delocalization and resonance energy or stabilize the TS. How does it happen? I don’t understand. I really need to know specifics because our professor gives us essay questions on the exam. I would really appreciate it.
1 answer
Sun Oct 28, 2012 10:34 AM
Post by amina gangat on October 22, 2012
Isn't it called aryl, not vinyl when it's sp2 and attached to a benzene ring?
1 answer
Sat Oct 20, 2012 10:02 AM
Post by singtong saipin on October 20, 2012
my bad its CH3CH2I+CH3CH2o^- ---->?NU: product
1 answer
Fri Oct 12, 2012 8:27 PM
Post by sophia lin on October 11, 2012
around the point 37:72 for the benzane questions You said that there is no rxn because the sp2 hybirdize carbon ..
but in the lecture about TS you said that the carbon is sp2 hybirize..if it has sp2 hybrize TS how come it does not have rxn?
1 answer
Tue Apr 17, 2012 11:04 AM
Post by Michelle Gavin on April 13, 2012
Can the slides be printed out for further study?
1 answer
Tue Mar 27, 2012 11:20 PM
Post by Pam Hillian on March 27, 2012
Under Substitutions at 17:27, the E vs. POR Diagram shows Starting Materials higher energy than Products, but audio states SN2 is Endothermic. The diagram drawn is Exothermic, isn't it? Please advise so I understand.
1 answer
Tue Mar 27, 2012 11:23 PM
Post by william le on March 8, 2012
thank you
1 answer
Fri Feb 24, 2012 11:49 AM
Post by Alibek Issabekov on February 23, 2012
Awesome! Terrific! Would have taken much more time, if I have read the book myself. Thanks a lot!
1 answer
Tue Dec 13, 2011 7:08 PM
Post by Jason Jarduck on December 10, 2011
Hi
I like your lectures because they help alot for preparing for exams.
Thank You
Jason Jarduck
1 answer
Tue Dec 13, 2011 7:09 PM
Post by Jason Jarduck on December 10, 2011
Hi Dr.Laurie Starkey,
I have a question C8H16 NMR with singlet, triplet, quintet and sextet.
I use the formula 2(8) + 2 =18 thus two missing hydrogens. One double bond.
I'm seem to always be getting the wrong structure such as one with a septet or a high number of singlets.
Also, a benzene ring with a primary alcohol group with reagents 1. SOCl2, pyridine 2. Li+ ethyne what would be the major product?
Thank you
Jason Jarduck
1 answer
Sun Nov 20, 2011 9:17 AM
Post by Karen O'Reilly on November 10, 2011
Thanks, Professor, exactly what I needed to know and "no arrow in the reaction flask", very funny :)
2 answers
Thu Nov 10, 2011 11:10 AM
Post by Karen O'Reilly on November 9, 2011
When a reaction arrow is in place and there are two items listed, a solvent and a Nu:, is the Nu: always listed on the top portion of the reaction arrow and the solvent listed underneath? For example, +Na-I over CH3OH
1 answer
Sat Nov 5, 2011 3:16 PM
Post by JUNCHAO ZHANG on November 1, 2011
i love your lectures!
TEST tomorrow! wish me luck! :)
1 answer
Fri Oct 12, 2012 11:15 PM
Post by sonia Orjuela Orjuela on October 13, 2011
Professor Starkey,
Thank you so much! you make organic chemistry to easy to understand. You should teach at Rutgers. =]
1 answer
Wed Aug 17, 2011 3:41 PM
Post by alex koralewski on August 8, 2011
In the synthesis slide (near the end), instead of using TsCl, could you (if it exists) use TsI ? Because then if you used TsCl, couldn't you end up with butyl chloride instead of the desired butyl iodide ?
1 answer
Sat Jul 30, 2011 12:23 AM
Post by Linda Wallace on April 23, 2011
I am trying to start at SN1 Mechanism section at 69.13. I do not need to watch the entire Substitution (108 minutes)section. Each time that I click on SN1 Mechanism it always go back to the beginning of Substitution.